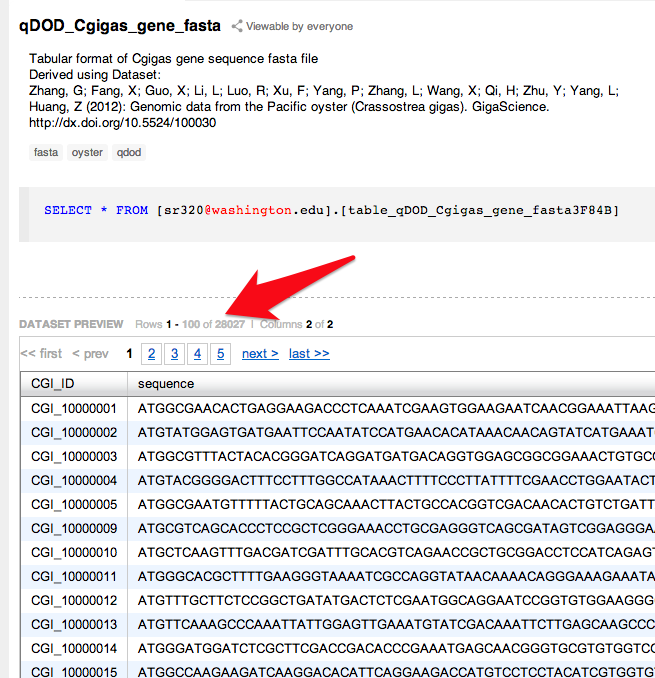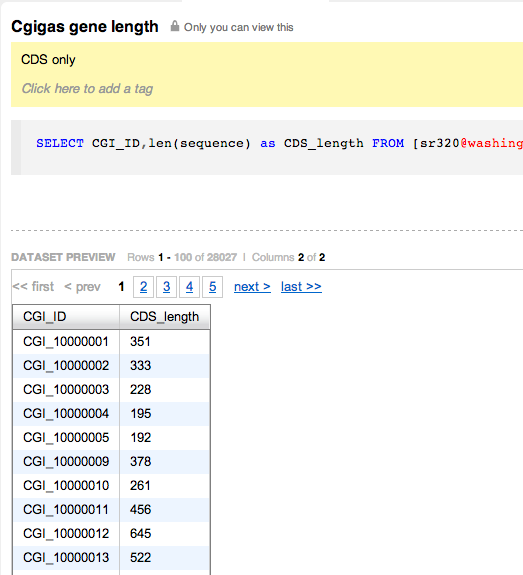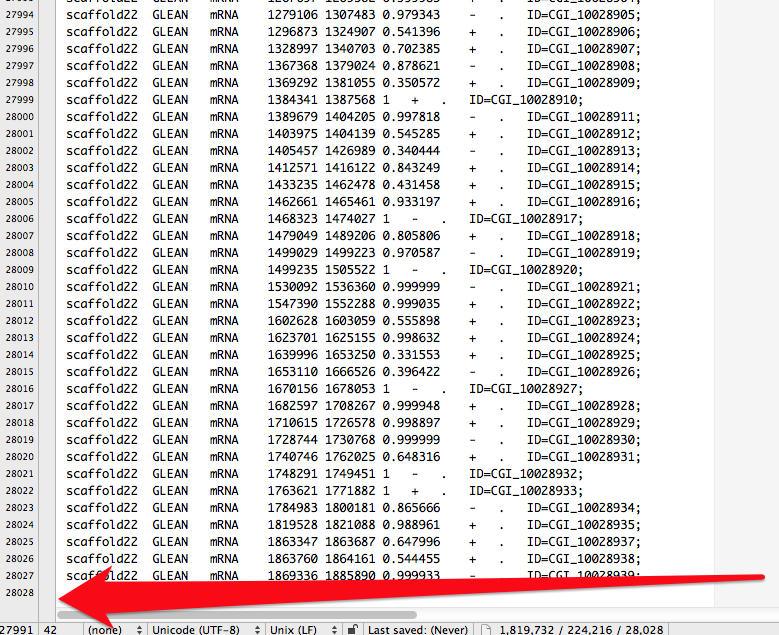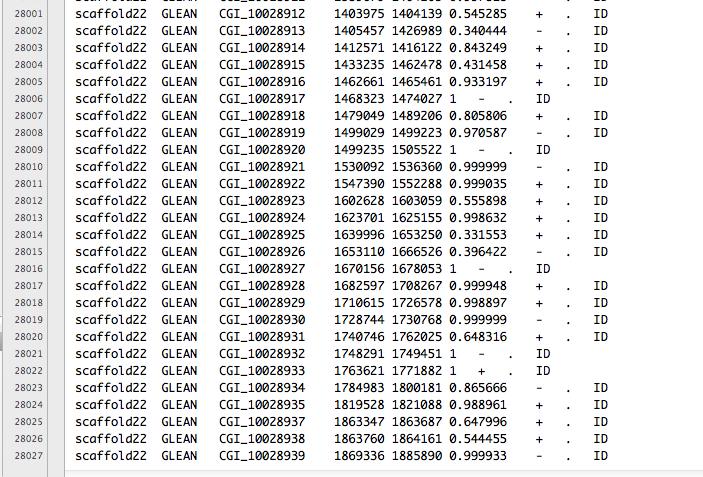
d-128-95-149-219:bsmap-2.74 sr320$ ./bsmap -w 1000 -a /Volumes/NGS\ Drive/NGS\ Raw\ Data/Oyster_BSseq/filtered_Unlabeled_NoIndex_L003_R1.fastq.gz -d /Volumes/web/cnidarian/TJGR_RepProMask_TE.fa -o /Volumes/web/cnidarian/BiGill_BSMAP_TEonly_v2.sam -p 16
BSMAP v2.74
Start at:FriJun1413:19:062013
Input reference file:/Volumes/web/cnidarian/TJGR_RepProMask_TE.fa (format: FASTA)
Loadin119787 db seqs, total size 39414569 bp.2 secs passed
total_kmers:43046721
Create seed table.5 secs passed
max number of mismatches: read_length *8% max gap size:0
kmer cut-off ratio:5e-07
max multi-hits:1000 max Ns:5 seed size:16 index interval:4
quality cutoff:0base quality char:'!'
min fragment size:28 max fragemt size:500
start from read #1 end at read #4294967295
additional alignment: T in reads => C in reference
mapping strand:++,-+
Single-end alignment(16 threads)
Input read file:/Volumes/NGS Drive/NGS RawData/Oyster_BSseq/filtered_Unlabeled_NoIndex_L003_R1.fastq.gz (format: gzipped FASTQ)
Output file:/Volumes/web/cnidarian/BiGill_BSMAP_TEonly_v2.sam (format: SAM)
BSMAP complete on Hummingbird
Total number of aligned reads: 2545683 (1.8%) Done. Finished at Sat Jun 15 20:45:07 2013 Total time consumed: 113161 secs
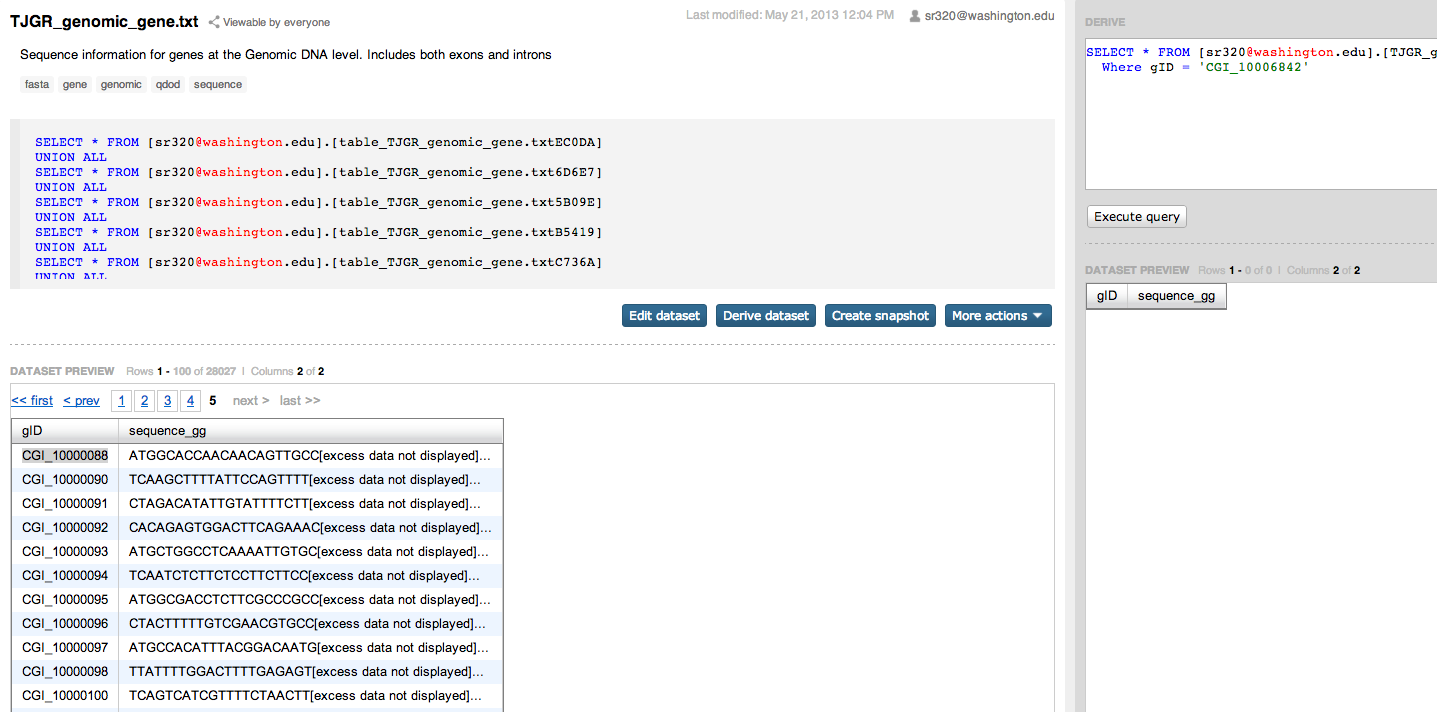
BiGill: Figuring out this Effective CT factor |
7.1 methratio.py
python script to extract methylation ratios from BSMAP mapping results. Require python 2.X.
For human genome, methratio.py needs ~26GB memory.
For systems with limited memory, user can set the -c/—chr option to process specified chromosomes only,
and combine results for all chromosomes afterwards.
Usage: python methratio.py [options] BSMAP_MAPPING_FILES
BSMAP_MAPPING_FILES could be one or more output files from BSMAP.
The format will be determined by the filename suffix.
(SAM format for *.sam and *.bam, BSP format for other filenames.)
Options:
-h, —help show this help message and exit
-o FILE, —out=FILE output file name. (required)
-d FILE, —ref=FILE reference genome fasta file. (required)
-c CHR, —chr=CHR process only specified chromosomes. [default: all]
example: —chr=chr1,chr2 (this uses ~4.5GB compared with ~26GB for the whole genome)
-s PATH, —sam-path=PATH
path to samtools. [default: none]
-u, —unique process only unique mappings/pairs.
-p, —pair process only properly paired mappings.
-z, —zero-meth report loci with zero methylation ratios.
-q, —quiet don’t print progress on stderr.
-r, —remove-duplicate
remove duplicated mappings to reduce PCR bias.
(This option should not be used on RRBS data. For WGBS, sometimes
it’s hard to tell if duplicates are caused by PCR due to high seqeuncing depth.)
-t N, —trim-fillin=N
trim N fill-in nucleotides in DNA fragment end-repairing. [default:2]
(This option is only for pair-end mapping. For RRBS, N could be detetmined by the distance between
cuttings sites on forward and reverse strands. For WGBS, N is usually between 0~3.)
-g, —combine-CpG combine CpG methylaion ratio from both strands. [default: False]
-m FOLD, —min-depth=FOLD
report loci with sequencing depth>=FOLD. [default: 1]
-n, —no-header don’t print a header line
-i CT_SNP, —ct-snp=CT_SNP
how to handle CT SNP (“no-action”, “correct”, “skip”),
default: “correct”.
“correct”: correct the methylation ratio according to the C/T SNP information
estimated by the G/A counts on reverse strand, see the output format below for details.
“skip”: do not report loci with C/T SNP detected (i.e. detected A on reverse strand)
“no-action”: do not consider C/T SNP.
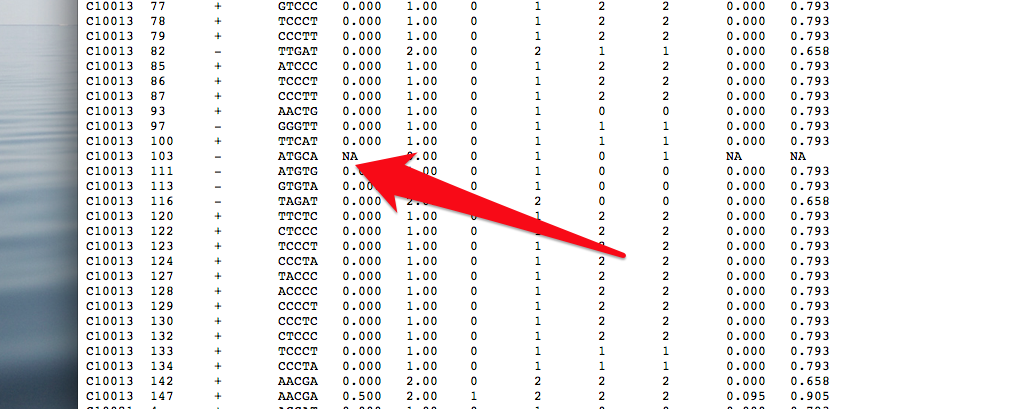
Output format: tab delimited txt file with the following columns:
1) chromorome
2) coordinate (1-based)
3) strand
4) sequence context (2nt upstream to 2nt downstream in Watson strand direction)
5) methylation ratio, calculated as #C_counts / #eff_CT_counts
6) number of effective total C+T counts on this locus (#eff_CT_counts)
CT_SNP=”no action”, #eff_CT_counts = #CT_counts
CT_SNP=”correct”, #eff_CT_counts = #CT_counts * (#rev_G_counts / #rev_GA_counts)
7) number of total C counts on this locus (#C_counts)
8) number of total C+T counts on this locuso (#CT_counts)
9) number of total G counts on this locus of reverse strand (#rev_G_counts)
10) number of total G+A counts on this locus of reverse strand (#rev_GA_counts)
11) lower bound of 95% confidence interval of methylation ratio, calculated by Wilson score interval for binomial proportion.
12) upper bound of 95% confidence interval of methylation ratio, calculated by Wilson score interval for binomial proportion.
Example:
python methratio.py —chr=chr1,chr2 —ref=hg19.fa —out=methratio.txt rrbsmap_sample*.sam
python methratio.py -d mm9.fa -o meth.txt -p bsmap_sample1.bsp bsmap_sample2.sam bsmap_sample3.bam
python methratio.py -s /home/tools/samtools -t 1 -d arab.fa -o meth.txt bsmap_sample.sam
Note: For overlapping paired hits, nucleotides in the overlapped part should be counted only once instead of twice.
methratio.py can correctly handle such cases for SAM format output, but for BSP format it will still be counted twice,
because the BSP format does not contain mapping information of the mate.
@1 day ago
BiGill - Gene Specific Methylation (Take 3) |
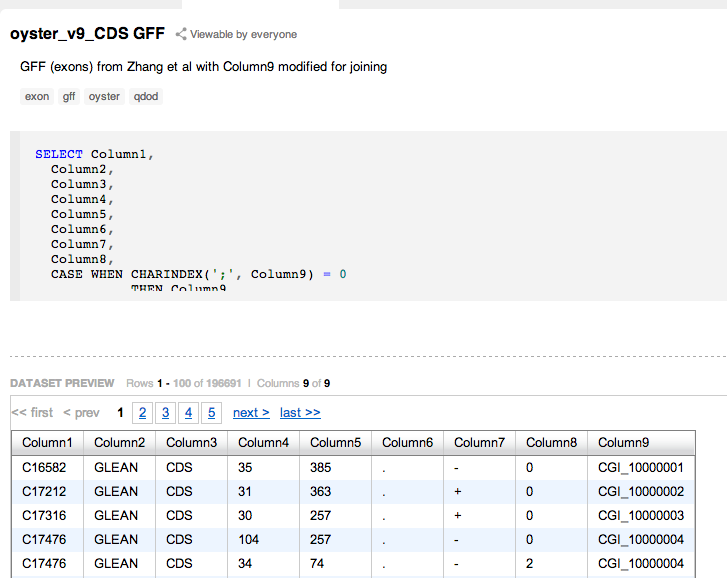
BiGill - Redoing Gene Specific Data |